
11-cis-retinal with Lys296 residue
(click to enlarge; 40kB)
Faculty of Chemistry, University of Gdansk, Sobieskiego 18, 80-952 Gdansk, Poland
Introduction
Bovine rhodopsin is a member of the largest G protein-coupled receptors (GPCR) subfamily. It consists of the protein (opsin; 348 amino acid residues) and 11-cis-retinal covalently linked to it through Lys296 residue. It is activated by light and turns on the signaling pathway that leads to vision. Absorption of a photon by 11-cis-retinal causes its isomerization to the all-trans form which, consequently leads to the conformational change of the protein moiety. After activating opsin all-trans-retinal is hydrolyzed and dissociated from it. The process can be repeated after binding newly synthesized 11-cis-retinal.
Methods
Recently published crystallographic structure of the inactive form of bovine rhodopsin (Palczewski, K., Kumasaka, T., Hori, T., Behnke, C.A., Motoshima, H., Fox, B.A., Le Trong I., Teller, D.C., Okada, T., Stenkamp, R.E., Yamamoto, M. and Miyamoto M.,Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor, Science 289, 739-745, 2000), called dark rhodopsin, was used as a start point in our computer-aided simulations. The main goal of this simulations was to check whether isomerization of 11-cis-retinal covalently linked to Lys296 into all-trans-retinal will cause considerable changes in conformation of the whole protein moiety, especially in the relative orientation of transmembrane helices. Parameterization of non-standard retinal residue, as well as minimization and relaxation of dark rhodopsin has been done in AMBER 5.0 suite of programs. We carried out all those simulations in vacuo.
Tools and procedures
Parameterization of non-standard retinal residue involved use of both Gamess98 and Amber5.0 computer programs. Geometry of retinal has been prepared based on crystallographic structure published by Palczewski et al. To make it readable by Amber suite of programs we had to describe three new types of atoms and double C-C bond - unknown in standard Amber database. This was made by approximation of relatively similar single and aromatic bonds existing in Amber database. The Gamess program has been used for generation of partial charges using quantum mechanics algorithms thereafter converted into atomic charges independent of conformation of retinal - this has been done in Amber (RESP module). Prepared retinal residue has been tested in vacuo, without receptor. These tests included standard constrained simulated annealing (CSA) procedures and change in value of cis-torsion (C11-C12) using minimization algorithms (both in Sander - an Amber module).
11-cis-retinal with Lys296 residue
(click to enlarge; 40kB)
Minimization and relaxation of dark-rhodopsin with previously prepared K296-retinal residue has been driven using starting model based on published crystallographic structure (Palczewski et all). In this part of our work standard minimization procedures has been used. Relaxation of the retinal/opsin complex has been joined with its initial molecular dynamics (MD) - applying this procedure we were able to get rid of most clashes in our structure.
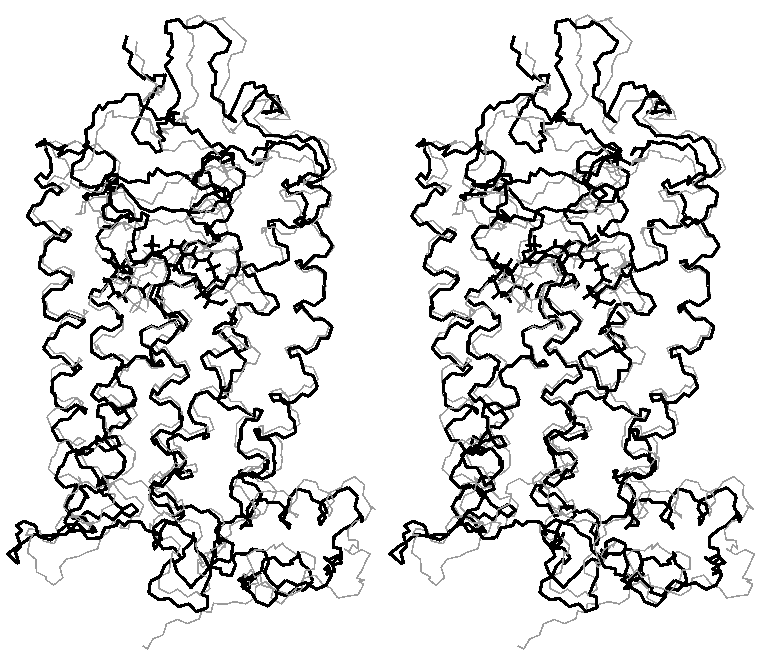
Relaxation of rhodopsin (stereo view); gray structure - structure before relaxation; black - after relaxation;
(click to enlarge; 32kB)

RMS changes during relaxation of rhodopsin
(click to enlarge; 6kB)
Molecular dynamics simulation of the cis-to-trans conversion of the TM7 K296-retinal C11-C12 bond has been the next step and the main goal of our work. This has been done using force constrains defining this torsion angle, changed 10 degrees every 8.5 ps of MD simulation. During this part of simulations the protein moiety should be deformed as it is necessary to allow changes in retinal conformation. After full change of C11-C12 torsion angle we carried further MD with final relaxation of this complex. Final minimization has been done after 198ps of MD simulations.
The isomerisation has beed simulated in two different ways: with C11-C12 torsion angle changes clock- and counterclockwise. These two "ways" lead us into two slightly different results - both presented on graphics below.
Superposition of rhodopsin structure before (77kB) and after (75kB) MD with isomerization of retinal - clockwise rotation
Structures colors: red - before rotation; blue - after rotation
(click to enlarge; 133kB)

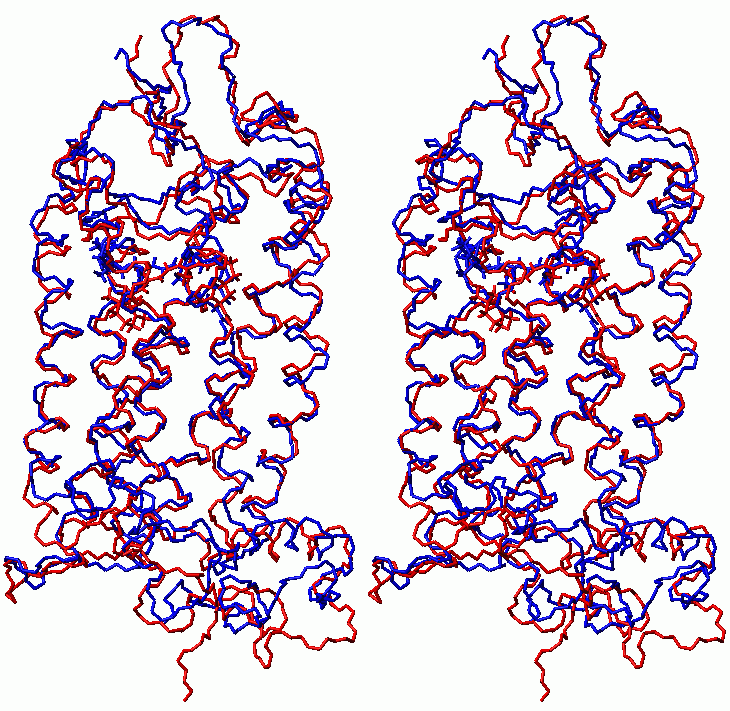
Superposition of rhodopsin structure before (73kB) and after (75kB) MD with isomerization of retinal - counterclockwise rotation
Structures colors: red - before rotation; blue - after rotation
click to enlarge; 130kB)

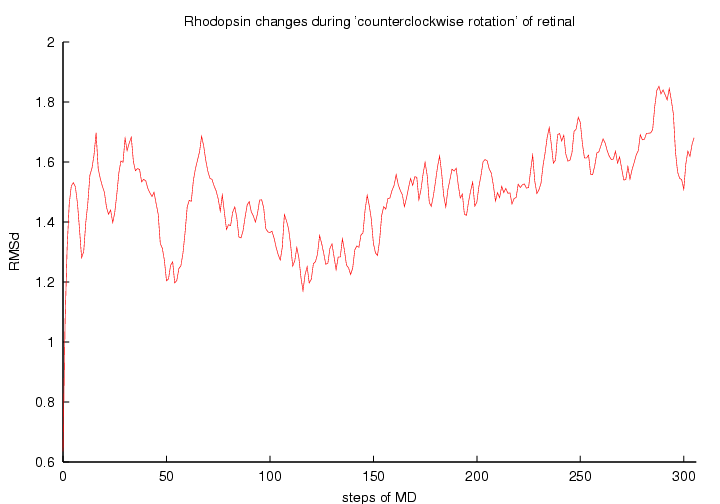
RMSd changes in conformation of rhodopsin in MD with 11-cis to all-trans retinal isomerisation: clockwise (left panell) and counterclockwise (right panell) rotation
(click to enlarge; 13 kB and 12 kB)
Results
Cis-to-trans conversion of retinal residue, hoped to be a driver of the RD change from the inactive/dark to the active/meta-II form, has failed in the respect that the resultant strain-less retinal trans isomer has brought about no major change in the protein shape.
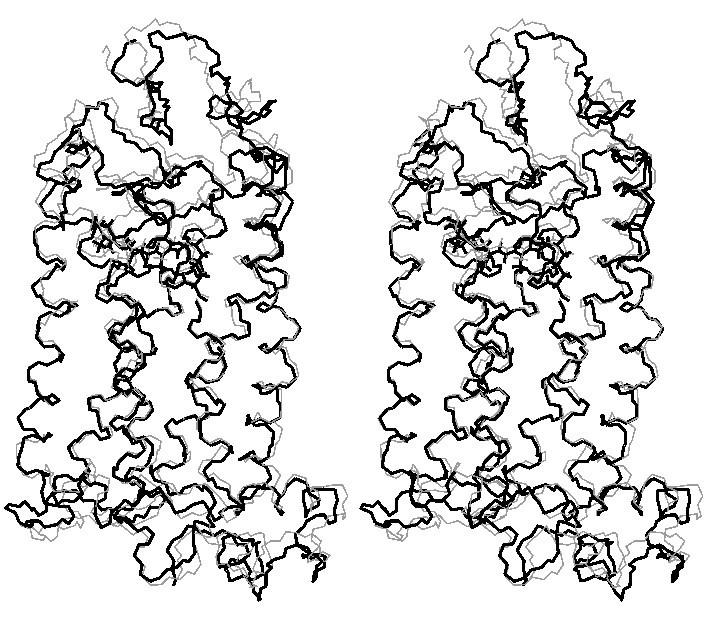
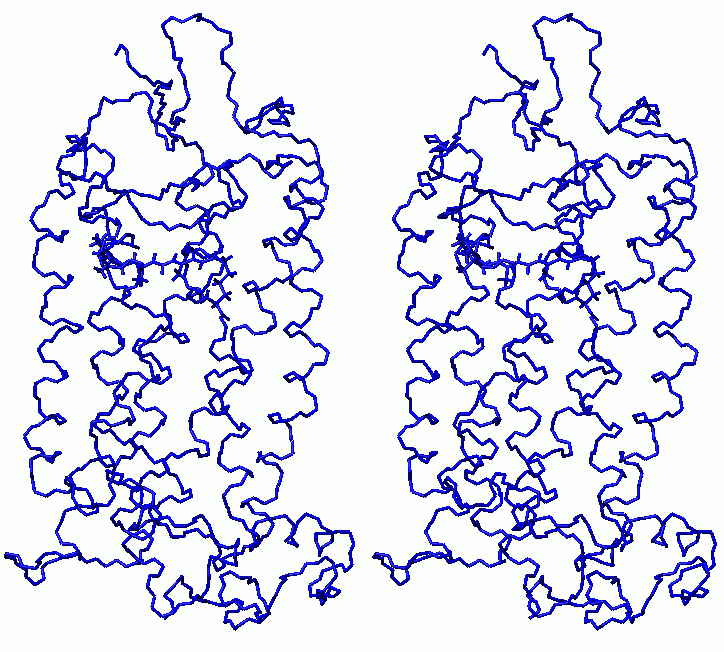
Final relaxation of rhodopsin (stereo view)
(gray - structure after molecular dynamics with clockwise rotation of C11-C12 retinal bond; black - final structure)
(click to enlarge; 28 kB)
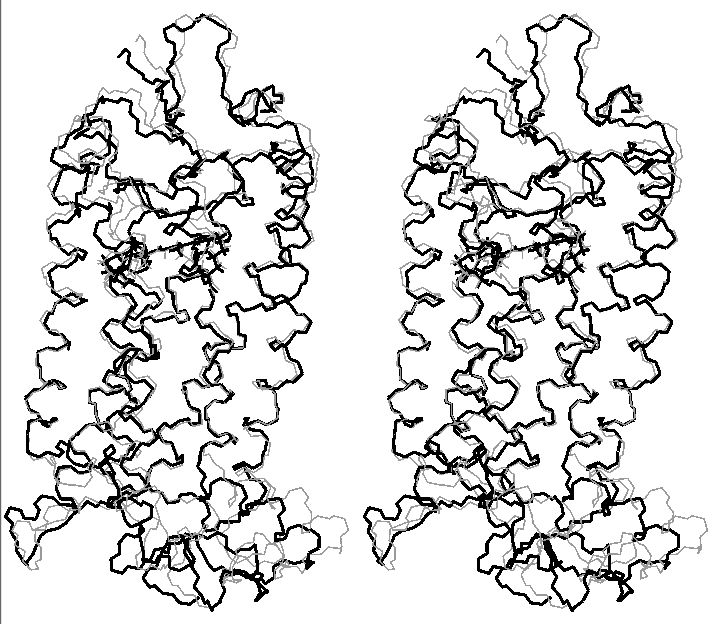
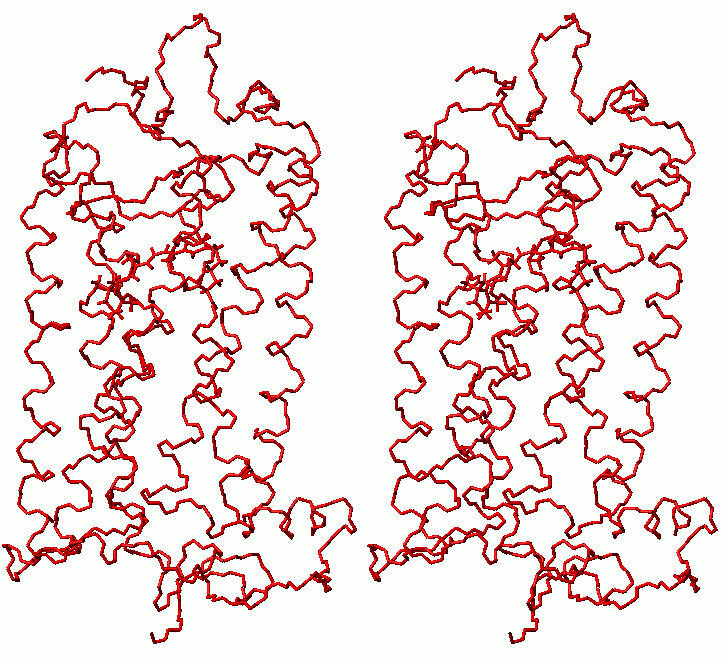
Final relaxation of rhodopsin (stereo view)
(gray - structure after molecular dynamics with clockwise rotation of C11-C12 retinal bond; black - final structure)
(click to enlarge; 31 kB)

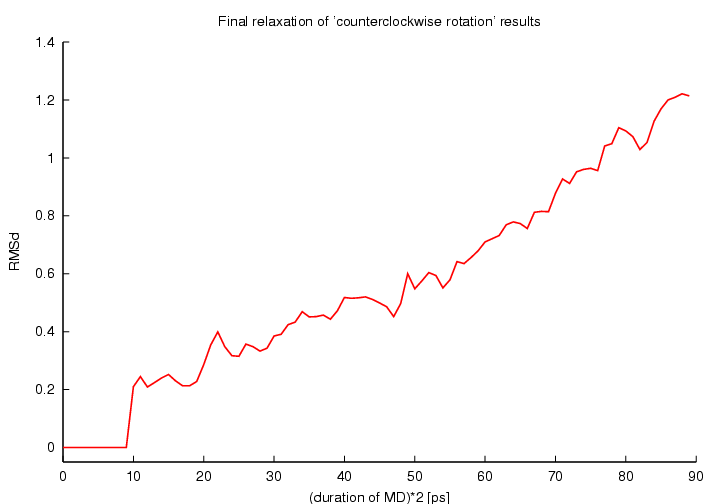
RMSd changes during final relaxations: structure after CW rotation (left panell) and after CCW rotation (right panell)
(click to enlarge; 2x8 kB)
A reason for this could be a too short (of total ~400 ps) simulations in relation to a very large protein-to-ligand inertia ratio. Thus, the next step is driving much longer simulations in lipid membrane including water molecules. Such a calculations should be much more realistic representation of changes in rhodopsin.

Superposition of rhodopsin before (gray) and after (black) molecular dynamics with clockwise rotation of C11-C12 retinal bond and final relaxation (stereo view)
(click to enlarge; 32 kB)
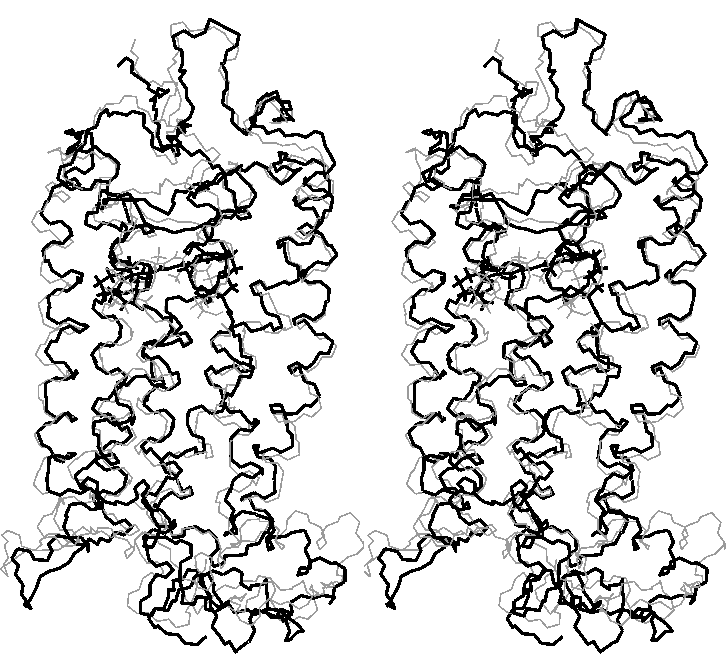
Superposition of rhodopsin before (gray) and after (black) molecular dynamics with clockwise rotation of C11-C12 retinal bond and final relaxation (stereo view)
(click to enlarge; 32 kB)
Used tools
Hardware:
Software:
{kind=link}
{kind=link}
{kind=link}
{kind=link}