Celem ćwiczenia jest opanowanie umiejętności przetwarzania plików graficznych z zastosowaniem programu Gimp.
Gimp, grafika, przetwarzanie obrazów
Na dysku Google uwtórz katalog tiwc11, który później udostepnisz prowadzącemu.
-
Wprowadzenie
Gimp jest przeznaczony do tworzenia i przetwarzania plików graficznych. Program Gimp jest programem darmowym i można go pobrać ze strony http://www.gimp.org.
-
Przygotowanie do ćwiczenia
Aby wykonać to ćwiczenie według podanej poniżej instrukcji, pobierz i zainstaluj program Gimp.
Jeżeli jednak masz juz zainstalowany dowolny inny program służący do edycji grafiki rastrowej (np. Adobe Photoshop), to nie musisz koniecznie instalować Gimp-a. Wykonaj poniższe polecenia w swoim programie w sposób możliwie najbliżej oddający oczekiwany efekt.
Strona z pakietami instalacyjnymi: https://www.gimp.org/downloads/ zawiera pakiety na większość popularnych systemów i architektur - wybierz odpowiednią dla Ciebie.
-
Uruchamianie programu Gimp
Program uruchom z menu, lub wpisując odpowiednią nazwę w dialogu uruchamiania (ALT-F2):
W celu uruchomienia programu w polskiej wersji językowej w pracowni TI (jeżeli z menu graficznego nie uruchomi się polska wersja) - należy w terminalu wpisać polecenie:
setenv LANG pl_PL
uruchomić program Gimp w terminalu tekstowym pisząc po prostu gimp i nacisnąć ENTER.
Na ekranie (po oknie powitalnym) pojawi się okno główne programu:

Ustaw najwygodniejszy dla siebie styl ikon i okna programu Gimp. -
Skalowanie rysunków
Pobierz ten plik
i zapisz go na dysku lokalnym.
Kliknij prawym przyciskiem na "czystej" części okna głównego i wybierz pozycję Plik a następnie opcję Otwórz. Otwiera się nowe menu z którego wybieramy zapisany przed chwilą plik dunes.jpg. Otwiera się dodatkowe okno zawierające plik z grafiką:
Gimp automatycznie zmienia rozmiar rysunku w taki sposób aby cały rysunek mieścił się w otwieranym oknie. W dolnej części okienka z rysunkiem znajdziemy podstawowe informacje o pliku, który właśnie został otwarty. Podana jest wielkość pliku (4.02MB), czy rysunek w czasie wyświetlania został zmniejszony lub zwiększony aby zmieścił się w całości w oknie(w naszym przypadku program musiał zmniejszyć rysunek o 66% w relacji do oryginalnych rozmiarów). Możemy sami zmieniać wielkość rysunku do czego służą przyciski bezpośrednio przy liczbie pokazującej aktualne zmniejszenie/zwiększenie rysunku. Okno w którym otwarty został rysunek otacza skala, która pozwala na określenie rozmiaru. W tym momencie wielkość rysunku wyrażona jest w postaci liczby tzw. pikseli określających wysokość i szerokość rysunku. Możemy zmienić tę skalę na np. skalę wyrażoną w milimetrach i do tego służy seria dwu przycisków obok symbolu px:

Często zdarza się, że musimy zmienić rozmiar rysunku. Może się tak dziać z wielu powodów: rysunek musi mieć odpowiednią wielkość wyrażoną np. w centymetrach lub calach aby ładnie mieścił się np. na stronie WWW, albo potrzebujemy rysunku o odpowiedniej wielkości pliku wyrażonego np. w MB czy kB aby mieścił się w e-mailu, albo potrzebujemy pliku o odpowiednich rozmiarach wyrażonych w pikselach aby np. mógł służyć jako tapeta w telefonie komórkowym. Manewrowanie rozmiarami pliku jest bardzo delikatnym zadaniem, bo w wyniku prostego zmniejszania, lub zwiekszania możemy utracić proporcje rysunku (zniekształcić go), jego rozdzielczość a czasami nawet i kolory. Gimp oferuje bardzo duże możliwości w manewrowaniu rozmiarami pliku. Aby zmienić rozmiary rysunku wybieramy z menu okna w którym jest otwarty rysunek Obraz a następnie z rozwiniętego menu opcję Skaluj obraz. Pojawia się nowe okno z opcjami skalowania obrazu:
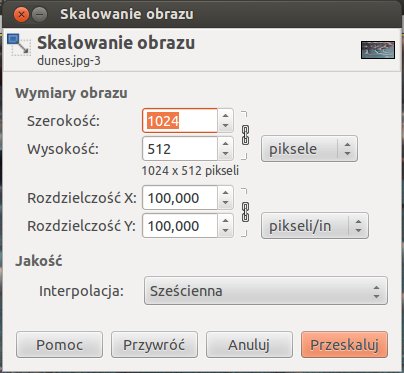
Możemy w tym menu operować wielkością rysunku, a wielkość może być wyrażona w różnych jednostkach w naszym przypadku wielkość obrazu jest wyrażona w pikselach (możemy to zmienić). Załóżmy, że chcemy obrazek który mamy powiększyć z obecnej szerokości 1024 piksele do szerokości 2000 pikseli. W tym celu w polu Szerokość wpisujemy liczbę 2000 i naciskamy ENTER. Proszę zauważyć, że po zmianie szerokości automatycznie zmieniła się też Wysokość tak aby rysunek zmienił się proporcjonalnie. Wysokość i szerokość są ze sobą związane o czym świadczy mały znaczek łańcuszka rozpietego pomiedzy nimi (można tę zależność zlikwidować). Po zmianie rozmiarów w opcji Interpolacja zmianiamy Sześcienna na Brak i naciskamy przycisk Skaluj. Jeżeli przyjrzelibyśmy się rysunkowi w dużym powiększeniu, powinniśmy zaobserwować podobny efekt:

Proszę odnaleźć jaka jest wielkość tego rysunku w MB? Wykonane przez nas skalowanie było skalowaniem prostym. Zmieniliśmy rozmiar rysunku więc program musiał "dodać" nowe piksele do rysunku aby go powiększyć. Dodawanie pikseli odbywało się na zasadzie ich "podziału". Każdy nowy piksel odziedziczył dokładnie taki sam kolor jak jego "przodek". Widać, że rysunek wyrażnie przybrał "ziarnistą" strukturę jak temu zaradzić? Wykonajmy jeszcze jedno skalowanie przy pomocy głównego menu wczytujemy jeszcze raz plik dunes.jpg. Otwieramy menu Obraz wybieramy opcję Skaluj, wpisujemy nową wysokość 2000 pikseli naciskamy ENTER a z menu Interpolacja wybieramy opcję Sześcienna i naciskamy przycisk Skaluj. Dostajemy nowy wariant naszego obrazka:

Czym się one różnią między sobą? Tym razem program też "wytworzył" nowe piksele ale tym razem nowe piksele zostały pokolorowane "wypadkową" kolorów pikseli z nimi sąsiadujących. Pokazano jak rysunki powiększać ale w podoby sposób odbywa się też ich zminiejszanie. Wyprodukowane rysunki są bardzo duże więc nie będziemy ich zapisywać, wybieramy z menu Plik następnie opcję Zamknij pojawia się pytanie w którym wybieramy opcję Nie zapisuj. To samo powtarzamy dla drugiego rysunku.
-
Kadrowanie
Czasami potrzebujemy z całego rysunku jedynie jego fragment. Za pomocą Gimpa można wykonać taką operację nazywaną kadrowaniem. W tym celu jeszcze raz wczytujemy rysunek dunes.jpg. Z menu głównego programu wybieramy opcję Kadrowanie:
kursor zmienia się w mały rysunek skalpela. Klikamy w jakiś punkt na rysunku z którego chcemy wyciąć mniejszy fragment i przeciągając myszką po rysunku zaznaczamy pole obszar rysunku który nas interesuje:

a następnie klikamy wewnątrz zaznaczonego obszaru. W miejsce oryginalnego rysunku pojawia się teraz tylko zaznaczony przez nas fragment:

Aby go teraz zapisać wybieramy Plik a następnie opcję Wyeksportuj jako. W nowym oknie wpisujemy w polu Nazwa dunes-maly.jpg i naciskamy Zapisz. Pojawia się jeszcze jedno okno w którym wybieramy OK (akceptujemy domyślne opcje). Gimp zapisuje pliki w bardzo wielu formatach, my wybraliśmy format JPG ale możemy wybrać inny używając innego rozszerzenia. Czy potrafisz otworzyć nowoutworzony plik za pomocą Gimp-a?
Zapisany przed chwilą plik dunes-maly.jpg prześlij na swój dysk Google do katalogu tiwc11 -
Zrzuty ekranu
Dość często zdarza się, że mamy coś na ekranie (pojedyncze okno, cała zawartość ekranu) co chcielibyśmy zachować. Przy pomocy Gimpa jest to bardzo proste. Z menu głównego wybieramy Plik a następnie opcję Utwórz i opcję Zrzut Ekranu.
Pojawia się nowe okno:

Jeśli wybierzemy opcję Pojedyncze Okno naciśniemy klawisz Zrzut i klikniemy na dowolne okno otwate na ekranie to Gimp otworzy nowe okienko z zachowanym obrazem. Jeśli wybierzemy opcję Cały ekran to po chwili otworzy się okno Gimpa z "fotografią" całego ekranu, co może wyglądać tak:

.
-
Retusz fotografii
Obecnie tradycyjna fotografia oparta na fotochemii jest wypierana przez fotografię cyfrową. Często wykonując zdjęcia nie jesteśmy zadowoleni z uzyskanych efektów i pojawia się ochota aby takie zdjęcia trochę "poprawić". Proszę pobrać i wczytać do programu Gimp plik: fotka.jpg. Co nam się bardzo nie podoba na tym zdjęciu to efekt "czerwonych oczu" nagminie pojawiający się na zdjęciach wykonanych z użyciem lampy błyskowej i jakaś plamka pod prawym okiem:
Nasz retusz zaczniemy od oczu. Po pierwsze powiększymy tylko obszar który nas interesuje w tym celu z menu głównego wybieramy opcję powiększenie:

i zaznaczamy myszką interesujący nas obszar czego efekt będzie wyglądał tak:

Następnie z menu głównego wybieramy opcję zaznaczania:

i myszką zaznaczamy "czerwone oko":

(ważne jest, aby kończąc zaznaczanie trafić dokładnie w punkt, z którego zaczęliśmy - inaczej zaznaczanie się nie zakończy). Z menu głównego wybieramy opcję wyboru kolorów:

Pojawia się menu wyboru kolorów, które pozwala na mieszanie podstawowych kolorów: czerwonego (R) zielonego (G) i niebieskiego (B), tak aby uzyskać interesujący nas kombinację. Dodatkowo można wybrać nasycenie i odcień (wartości H,S,V). Musimy teraz dobrać jakiś kolor, który zastąpiłby "czerwone oko" czymś bardziej rzeczywistym (proszę popatrzeć jak wyglądają i mają kolor źrenice u sąsiadów). Tutaj wybrano następujący zestaw kolorów:

Po wyborze koloru z menu głównego wybieramy opcję wypełniania kolorem zaznaczonych obszarów:

a następnie klikamy na zaznaczony wcześniej obszar "czerwonego oka". Całą procedurę powtarzamy dla drugiego oka. Teraz zabieramy się za skazę pod prawym okiem (przy okazji widać jak wygląda "naprawione oko":

Aby zlikwidować przebarwienie musimy zorientować się jaki kolor ma normalna skóra w okolicy. W tym celu z menu głównego wybieramy opcję Pobierz kolor:

i klikamy w okolice plamki której chemy się pozbyć. Po kliknieciu "pipetką" na rysunku, w miejscu, gdzie podany jest aktualny kolor rysowania pojawia się wybrany właśnie kolor:

Teraz możemy przeprowadzić usuwanie przebarwienia. W tym celu z menu głównego wybieramy opcję pędzel:

W Gimpie można używać różnych pędzli my będziemy używać pędzla tzw. "Fuzzy" który mocno pokrywa miejsca gdzie pędzel dotyka malowanej powierzchni ale jednocześnie część farby "rozpryskuje się" na boki. Możemy regulować przy pomocy opcji Krycie jak mocno nasza farba jest rozcieńczona. Po wyborze farby i narzędzi malujemy, a efekt powinien być taki:

Nasze dzieło po ukończeniu powinno wyglądać jak na poniższym przykładzie:

Prześlij plik fotka-poprawiona.png na swój dysk Google, do katalogu tiwc11
-
Klonowanie tekstury
Niejednokrotnie przygotowując grafiki jesteśmy zmuszeni do wykonania różnego rodzaju fotomontaży. Gdy używamy standardowego pędzla, a nie mamy doświadczenia w tego typu edycji, zazwyczaj nasze poprawki są zbyt widoczne. Jak zatem można usunąć coś ze zdjęcia w subtelny sposób?
Bardzo przydatną opcją jest na przykład klonowanie tekstury.
Z menu wybierz narzędzie klonowania a następnie przytrzymując klawisz „CTRL” lewym klawiszem myszy zaznaczasz obszar do klonowania. Puszczamy „CTRL” i już możemy malować teksturą z obrazu.
Uwaga! Wraz z malowaniem - obszar kopiowany przemieszcza się.Wykonaj:
Używając klonowania spróbuj wymazać śmigło helikoptera widoczne na przykładzie poniżej.
Obrazek potrzebny do wykonania zadania zapisz na swoim dysku lokalnym bezpośrednio z przeglądarki (użyj prawego przycisku myszy):
Obraz, jaki powinieneś otrzymać po wykonaniu zadania, powinien wyglądać mniej więcej jak na poniższej grafice (bez napisu "wzór"... ;)
Wyeksportuj poprawiony obrazek helikoptera pod nazwą helikopter_bez_smigla.png Użyjesz go w kolejnym zadaniu.
-
Warstwy - wklejanie i modyfikacja.
Wykonaj:
Używając przeglądarki internetowej, znajdź dowolny obrazek butelki Coca-Coli. Zapisz obraz na dysku lokalnym.
Otwórz go i następnie używając narzędzia do odręcznego zaznaczania obszarów obrysuj dokładnie butelkę. Ważne jest, aby kończąc zaznaczanie trafić dokładnie w miejsce, z którego zaczęliśmy zaznaczanie, wówczas uzyskamy zamkniętą krzywą (pełne zaznaczenie).
Obrysowaną butelkę skopiuj (Ctrl+c), a następnie wklej (Ctrl+v) do obrazka z helikopterem bez śmigieł.
Aby dopasować wielkość wklejonej butelki, kliknij prawym klawiszem myszy i wybierz: Warstwa -> Skaluj Warstwę. Podobnie wklej pozostałe butelki. Poeksperymentuj z narzędziem do skalowania by uzyskać efekt podobny jak na poniższym rysunku:
Wyeksportuj gotowy obraz pod nazwą helikopter_cola.png
Gotowy obraz prześlij do katalogu tiwc11 na swoim dysku Google.
Ten sposób przenoszenia wycinka obrazu do innego jest dobry dla małych lub prostych obiektów. W przypadku potrzeby pozbycia się większej części obrazu, lub zdefiniowania przezroczystości zastosujemy za chwilę inna technikę. -
Przezroczystości
Zapisz z przeglądarki prezentowaną poniżej grafikę prostego dwucukru na niebieskim tle. Plik nazywa się struktura.jpg
Twoje zadanie polega na przygotowaniu tej grafiki w taki sposób, aby można ją było wkleić do prezentacji lub innego obrazka bez niebieskiego tła.
Jeżeli mamy do czynienia z prostą sytuacją, w której usunąć trzeba jednolite tło, w jednym, określonym kolorze, to najprościej jest to zrobić zamieniając ten kolor na specjalną warstwę definiującą przezroczystości. W grafice nazywa się ją "kanałem alfa". Zamień niebieskie tło na kanał alfa wybierając z menu (prawy przycisk myszy na obrazku) pozycje: "Warstwa > Przezroczystość > Zamiana koloru na alfę...", jak na grafice:

W nowym okienku kliknij na ramkę, w której zdefiniowany jest kolor, który będziemy zastępowali przezroczystością (numer "1" na kolejnej grafice), później kliknij narzędzie wyboru koloru ("pipetkę") - numer "2" i w końcu wskaż dowolny punkt na tle, które ma zostac wycięte (numer "3"):

Po zatwierdzeniu obydwu uruchomionych dialogów (przyciskami "OK") niebieski kolor z tła zniknie bez śladu...
Jeżeli zdarzy się, że nie cały niebieski kolor zostanie wycięty - wróć do dialogu wyboru koloru i zadbaj o to, aby wybrany kolor ("Zapis języka HTML") nazywał się dokładnie: 0000ff.
W miejscu koloru widzimy teraz kratkę/szachownicę. W ten sposób Gimp oznacza obszar, w którym nie został zdefiniowany żaden kolor:
Aby zachować taki obrazek do późniejszego wykorzystania musisz go zapisać w formacie, który umożliwia przechowywanie informacji o przezroczystości - format "jpeg" do takich nie należy. Najlepiej (bo bez straty jakości) zapisać swoja grafikę w formacie PNG (z rozszerzeniem "png"). Tylko wtedy możliwe będzie wklejenie takiej grafiki bez tła do prezentacji, dokumentu lub do innej grafiki.
Dodatkowa informacja - tę część możesz pominąć :)
Wklej swoją wyciętą z tła strukturę na tło zrobione z tego obrazka. Na połączonej grafice dodaj swoje imię zapisane w wybranym kolorze. Do imienia możesz też dodać cień ("Filtry > Światło i cień > Rzucanie cienia..."). Możesz osiągnąć wynik podobny do:
Jeżeli w jakimś pliku graficznym do "wycięcia" jest więcej, niż jeden kolor, to nie można prosto zamienić jednego, wybranego koloru na kanał alfa, ponieważ to nie wystarczy. W takim przypadku należy najpierw do pliku oddać kanał alfa ("Warstwa > Przezroczystość > Dodaj kanał alfa") - o ile kanału alfa jeszcze w nim nie było, a następnie dowolnym sposobem należy zaznaczyć dowolny fragment grafiki (narzędziami do zaznaczania) lub kolor (narzedziem do wyboru koloru) i go wyciąć (Ctrl-X).
Podczas wycinania (usuwania) fragmentu grafiki w sytuacji, kiedy został zdefiniowany kanał alfa powoduje, że wycięte obszary stają się przezroczyste (a nie w kolorze tła). Można tym sposobem usuwać z dowolnej grafiki dowolne elementy, zaś pozostałą część zapisać w formacie png - do wykorzystania w dowolnym momencie i celu.
Tworzenie przezroczystych obszarów zamiast tła w konkretnym kolorze jest szczególnie przydatne np. podczas tworzenia prezentacji multimedialnych, kiedy np. tło slajdu ma określony kolor, nizależny od wstawianej grafiki. O wiele ładniej wyglądałby sam kolorowy kwiatek na trawiastej łące, niż ten sam kwiatek na tle białego prostokąta wklejony na tę samą trawiastą łąkę...
Powstały obraz zapisz pod nazwą grafika.png i prześlij do katalogu tiwc11 na swoim dysku Google.
- Obróbka fotografii
Profesjonalnie odnawianie starych fotografii jest bardzo kosztowne. Używając darmowego GIMP’a możemy to zrobić samodzielnie, inwestując tylko trochę czasu i zapału.
Wykonaj:
Zapisz na dysku lokalnym widoczną poniżej starą fotografię (zrób to bezpośrednio klikając na poniższą grafikę prawym klawiszem myszy i wybierz Zapisz obraz jako...).
Poeksperymentuj z ustawieniem kolorów oraz dostępnymi filtrami a także z opcją pobierania koloru, aerografem i klonowaniem tekstury i popraw fotografię tak, by uzyskać jak najlepszy efekt (przynajmniej tak dobry jak poniżej).
Wykaż się inwencją i kreatywnością. Pozbądź się wszystkich plam i "dziur" w zdjęciu. Popraw kontrast i ostrość. Każda modyfikacja, która podniesie czytelność starej fotografii jest dobra...
Przykładowe zdjęcie po przeróbce:

Gotowy plik wyeksportuj pod nazwą foto2.pngPlik foto2.png prześlij na swój dysk Google, do katalogu tiwc11.
{kind=link}
{kind=link}
{kind=link}
Katalog tiwc11 na dysku Google udostępnij prowadzącemu (powinien zawierać 5 plików graficznych).