Rafał Ślusarz
Analiza wyników MD będzie polegała na ocenie zmian energetycznych i konformacyjnych wymodelowanych kompleksów.
Poniższe wskazówki dotyczą każdego z modelowanych kompleksów osobno (czyli wszystkie wskazane czynności należy powtórzyć cztery razy).
W katalogu, w którym wykonała się dynamika kompleksu, należy stworzyć dodatkowy podkatalog (np. o nazwie analiza) i to w nim należy wykonywać wszystkie opisane czynności, aby uniknąć bałaganu i nadpisywania plików.
- Przygotowanie plików danych
Najprościej jest zastosować standardowy skrypt do oddzielenia uzyskanych zmian układu z plików wyjściowych MD o nazwie process_mdout.perl. Jako argument podaje się mu nazwę pliku wyjściowego przebiegu MD (plik OUT). My dysponujemy kilkoma takimi plikami (z każdego przebiegu MD osobnym), więc należy wszystkie pliki wyjściowe połączyć w jeden plik (nie polecam) lub podać wszystkie nazwy tych plików razem (polecam):
process_mdout.perl ../*.out
W wyniku działania tego skryptu otrzymamy 33 pliki wynikowe, zapisane w katalogu bieżącym. - Wykresy zmian energii
Najbardziej interesują nas zmiany energii kinetycznej (summary.EKTOT), potencjalnej (summary.EPTOT) i całkowitej (summary.ETOT) i właśnie te wykresy muszą się znaleźć w pracy dyplomowej.
Dynamika była prowadzona w warunkacj stałego ciśnienia i temperatury, więc potwierdzeniem tego, że została wykonana poprawnie, mogą być wykresy sporządzone z plików summary.PRES oraz summary.TEMP i ich pochodna: summary.VOLUME.
W pracy muszą się też znaleźć wykresy wspólne, z nałożeniem wszystkich czterech przebiegów wybranej energii (całkowitej, potencjalnej lub kinetycznej - zależnie od tego, które z tych energii będą najlepiej różnicowały poszczególne kompleksy).
Wykresy można przygotować dowolną metodą i dowolnym zestawem narzędzi, należy jednak zwrócić uwagę na dwie rzeczy:- pełen wykres przygotowany z wyodrębnionych plików będzie zawsze zawierał niekorzystny, zaciemniający obraz zmian początek:
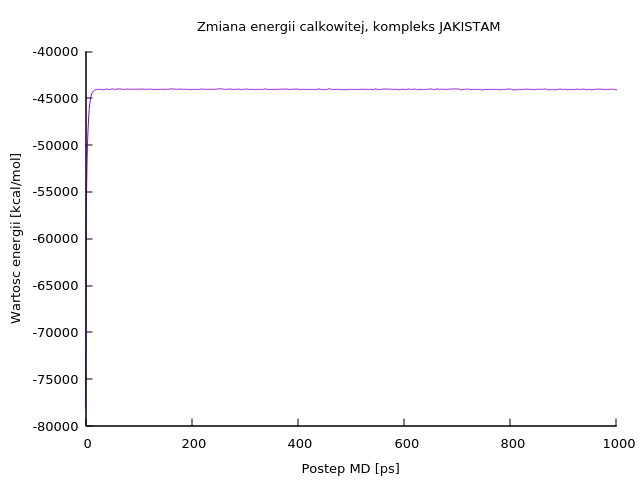 Jest to normalne (ze względu na początkowe dostosowywanie się układu), ale niepożądane. Możliwe są w tej sytuacji dwa rozwiązania:
Jest to normalne (ze względu na początkowe dostosowywanie się układu), ale niepożądane. Możliwe są w tej sytuacji dwa rozwiązania:
- pozostawienie wykresu w oryginalnej postaci i szerokie omówienie powodu wypłaszczenia wykresu i nieprzystającego do reszty jego początku
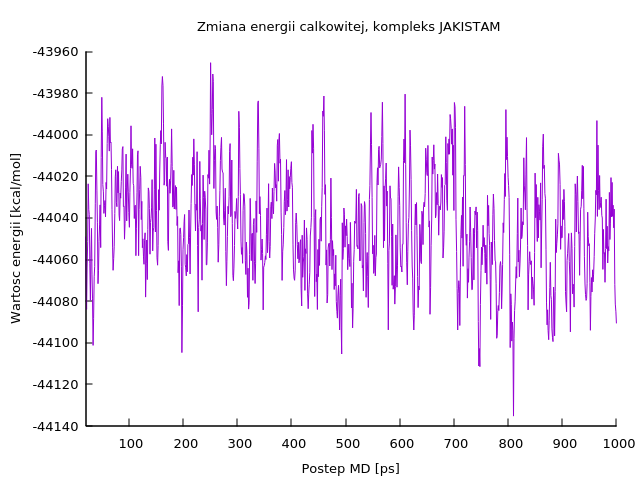
- obcięcie kilku początkowych kroków tak, aby zostawić tylko spójny przebieg. Poniżej znajduje się ten sam wykres, po obcięciu 30-tu pierwszych kroków:
- punktów pomiarowych jest 1000; podczas przygotowywania nałożonych na siebie wykresów energii dla poszczególnych kompleksów liczba ta może się okazać zbyt dużą - obraz trendów będzie nieczytelny, wykresy będą przykrywały same siebie:
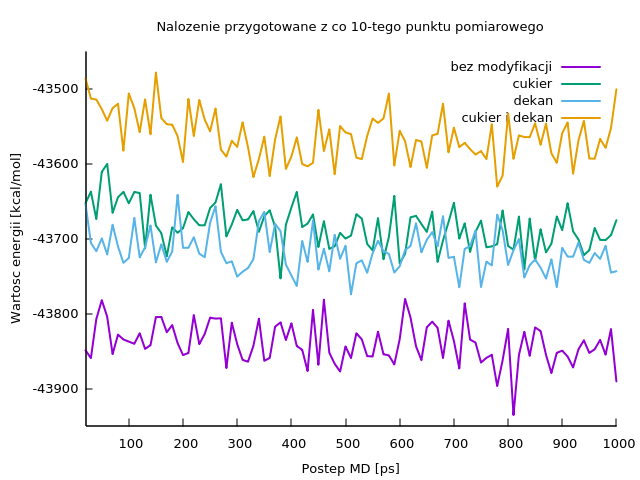
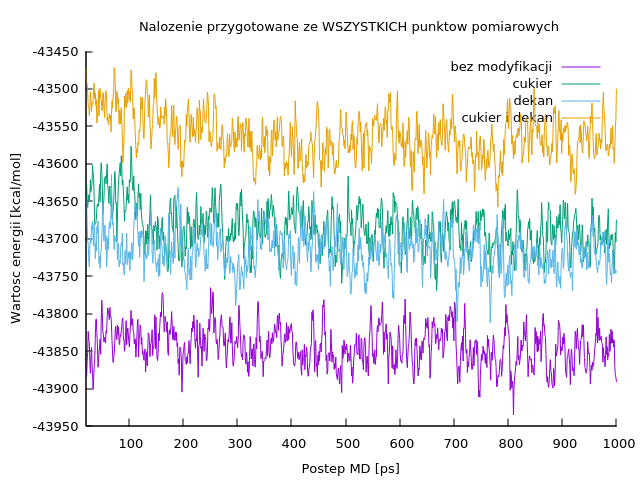 W takiej sytuacji należy sporządzić poszczególne linie wykorzystując nie wszystkie, tylko co 5-ty, co 10-ty lub co 20-ty punkt pomiarowy, na przykład:
W takiej sytuacji należy sporządzić poszczególne linie wykorzystując nie wszystkie, tylko co 5-ty, co 10-ty lub co 20-ty punkt pomiarowy, na przykład:
- pełen wykres przygotowany z wyodrębnionych plików będzie zawsze zawierał niekorzystny, zaciemniający obraz zmian początek:
- Pomiary zmian konformacyjnych
Pomiary te należy wykonać w narzędziu cpptraj, do którego potrzebny jest plik wejściowy cpptraj.in. Przykładowy plik cpptraj.in:
trajin ../00.crd trajin ../01.trj trajin ../02.trj trajin ../03.trj trajin ../04.trj strip :WAT rms first :1-7 out rms.out [wanko] rms first :8-9 out rms.out [cukier] rms first :10-15 out rms.out [pentapep] distance wod1 :15@O,OXT :2@H out odleglosci.out distance wod2 :15@O,OXT :3@H out odleglosci.out distance wod3 :15@O,OXT :4@H out odleglosci.out distance wod4 :15@H :4@O out odleglosci.out distance wod5 :13@O :7@H out odleglosci.out distance WanPP :1-7 :10-15 out odleglosci.out run
- trajin - ładowanie koordynatów (pierwszej klatki, lub całych trajektorii); należy wskazać ścieżkę i nazwy wszystkich uzyskanych trajektorii (w przykładzie, powyżej wypisane są tylko 4 przebiegi)
- strip - usuwamy z obrazu cząsteczki wody
- [w_nawiasie] - nagłówek kolumny z wynikami RMSd zapisany w pliku rms.out
- rms - wyliczenie odchylenia średniokwadratowego w stosunku do pierwszej klatki (first); wyniki pomiaru zostaną zapisane w pliku rms.out, zaś mierzyć odchylenie będziemy we fragmentach zdefiniowanych numerami (zakresem) reszt; w przykładzie powyżej są to trzy fragmenty: wankomycyna bez części cukrowej (aglikon wankomycyny), sama część cukrowa i sam pentapeptyd. Gdyby wankomycyna była podstawiona na C-końcu resztami o numerach 10 i 11, zaś część cukrowa miała dodatkowo dołączony dekan o numerze 12, to zakresy numerów reszt byłyby od siebie oddzielone przecinkiem:
rms first :1-7,:10-11 out rms.out [wanko] rms first :8-9,:12 out rms.out [cukier] rms first :13-17 out rms.out [pentapep]
UWAGA na spacje (lub ich brak!). W pierwszej linijce, powyżej, jest tylko pięć spacji; nie może być spacji pomiędzy ":1-7," a ":10-11" - distance - obliczenie odległości pomiędzy atomami lub środkami mas podanych zakresów atomów; np. :15@O,OXT :2@H oznacza odległość pomiędzy {środkiem masy wyznaczonym przez obydwa atomy tlenu grupy karboksylowej reszty o numerze 15} a {amidowym atomem wodoru należącym do grupy o numerze 2}. Zapis :15@H :4@O oznacza prostą odległość pomiędzy atomami "O" i "H" należącymi do reszt "15" i "4"; można też zdefiniować odległość grupy atomów od innej grupy atomów, definiując zakres reszt tworzących tę grupę, np. :1-7 :10-15 (odległość pomiędzy środkiem masy aglikonu wankomycyny a środkiem masy pentapeptydu, z pominięciem części cukrowej). W kompleksach złożonych ze zmodyfikowanego C-końca - należy uwzględnić reszty je tworzące dodając dodatkową linijkę odległości {C-koniec} do {pentapeptyd}, na przykład:
distance WanPP :1-7,:10-11 :12-16 out odleglosci.out
- jeżeli C-koniec tworzyłyby reszty o numerach 10 i 11, zaś pentapeptyd miał w tym układzie numery reszt z zakresu 12 do 16 (w kompleksie z dekanem lub bez niego te numery reszt będą oczywiście różne).
UWAGA na spacje (lub ich brak!). W linijce, powyżej, jest dokładnie pięć spacji; nie może być spacji pomiędzy ":1-7," a ":10-11", za to musi być spacja pomiędzy ":1-7,:10-1" a ":12-16".Słowo "WanPP" (podobnie, jak powyższe "wod1", "wod2" itd) są nagłówkami/identyfikatorami zbioru wyników i zostaną zapisane jako nagłówki kolumn w pliku wynikowym odleglosci.out. Dzięki tym nagłówkom, rysując wykres z wykorzystaniem kolumny 1 i 3 lub kolumn 1 i 6 będziemy wiedzieli, że rysujemy wykres "wiązania wodorowego 2" lub "wiązania wodorowego 5".
Pomijamy odległości od dekanu, ponieważ nie powinien on brać udziału w oddziaływaniu z pentapeptydem.
Obliczenia odległości pomiędzy pentapeptydem a wankomycyną oraz obliczenia długości wiązań wodorowych pomiędzy nimi są krytyczne dla Waszej pracy i najważniejsze ze wszystkich wyników. To na ich podstawie będziecie wnioskować, czy i jak modyfikacje struktury wankomycyny wpływają na wiązanie tego antybiotyku do C-końca peptydoglikanu, który reprezentuje tu ścianę komórkową bakterii.
Uruchomienie obliczenia:
cpptraj -p ../topologia < cpptraj.in
- Wykresy zmian konformacyjnych
Należy przygotować wykres zależności odległości środka masy aglikonu wankomycyny od pentapeptydu od czasu symulacji oraz wykresy obrazujące długości pięciu określonych badaniami strukturalnymi wiązań wodorowych - wszystkie 6 przebiegów najlepiej we wspólnym układzie współrzędnych, aby można było je porównywać i dyskutować o zmianach przebiegów. Przykładowy zbiór wykresów, przygotowany w Gnuplot-cie, z uwzględnieniem co 10-tego punktu pomiarowego (z pliku odleglosci.out), z liniami o grubości 2: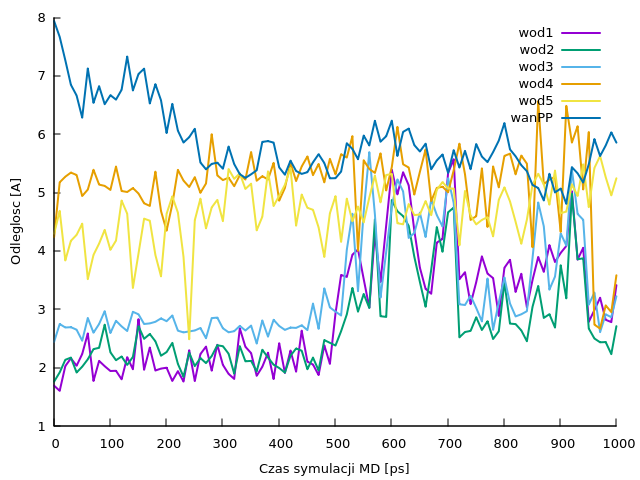
Więcej na temat prostego wykorzystania programu Gnuplot wprost na pracowni znajduje się na tej stronie.Należy również przedstawić zmiany RMSd poszczególnych fragmentów kompleksu w przebiegu MD (w każdym kompleksie osobno) ORAZ nałożyć na wspólny wykres wybrane zmiany RMSd aglikonu wankomycyny albo pentapeptydu ze wszystkich kompleksów - zależnie od tego, które zmiany lepiej będą różnicowały kompleksy. Można oczywiście wykonać więcej wykresów i więcej nałożeń, o ile uda się dzięki temu pokazać dodatkowe podobieństwa lub różnice w przebiegach MD dla poszczególnych kompleksów.
- Interpretacja zmian RMSd, energii i odległości
Energie: jak zawsze: im niższa energia, tym korzystniejsza - i z punktu widzenia oddziaływania pomiędzy cząsteczkami i całego układu.RMSd - im wyższa wartość, tym dalej idące zmiany w strukturze; konformacja struktury (lub jej fragmentu) mniej przypomina swoją konformację na początku symulacji lub jej konformacja wraca do (lub w kierunku) konformacji startowej - jeżeli wartość RMSd spada w trakcie symulacji. Nie ma tu pojęcia "dobrej" czy "złej" wartości; jest po prostu "inna" wartość.
Miarami RMSd najlepiej jest porównywać zmienność fragmentów kompleksu pomiędzy układami (czy np. zmiany RMSd są większe w przypadku kompleksu z obecnym dekanem (bo powinny być), czy wielkim C-końcem, itp.).Należy pamiętać, że odczytane z MD wartości energi odnoszą się do całych układów, z różną ilością cząsteczek wody i różnymi antybiotykami - nie można więc bezpośrednio porównać wartości energii jednego układu z wartością tej samej energii drugiego układu. Wolno nam za to porównać trend, zachowanie zmiany takiej energii.
Za to odległości i długości wiązań wodorowych (obydwie te wartości wyrażone w Angstromach) wolno nam porównywać bezpośrednio i bez ograniczeń - tak właśnie zróbcie. Wydłużenie obserwowanych wiązań wodorowych lub ich zerwanie (powiększenie powyżej ok. 5-ciu Å) świadczyć będzie o tym, że wprowadzona modyfikacja struktury wankomycyny zadziałała niekorzystanie na wiązanie wanko-pentap. Utrzymanie wszystkich pięciu wiązań wodorowych jest warunkiem koniecznym.
- Pozostałe wizualizacje i opisy
W pracy dyplomowej, jeszcze przed umieszczeniem i omawianiem jakichkolwiek wykresów, należy przedstawić pełnoatomowe reprezentacje:- samej wankomycyny, wankomycyny modyfikowanej (na dwa sposoby)
- pentapeptydu
- kompleksu wankomycyny z pentapeptydem (lub wankomycyny modyfikowanej z pentapeptydem) po dokowaniu i ostatniej minimalizacji
- pudła periodycznego zawierającego wszystkie składowe kompleksu, wodę i przeciwjony (tu wystarczy raczej jeden, wspólny rysunek, ze względu na mocno ograniczoną przejrzystość takiej reprezentacji (czyt.: woda zasłoni wszystko))
- kompleksów wankomycyna-pentapeptyd po dynamice
- dla ambitnych: nałożonych na siebie aglikonów wankomycyny; nałożenia można dokonać wczytując do PyMOL-a cztery pliki PDB pozbawione wody, przeciwjonów, części cukrowych i pentapeptydów, wykorzystując wewnątrz PyMOLa polecenie "fit":
fit obiekt2, obiekt1
fit obiekt3, obiekt1
- w ten sposób obiekty "2" i "3" zostaną nałożone/dopasowane do położenia obiektu "1". Po pokolorowaniu obiektów na wybrane przez siebie kolory i wyłączeniu wyświetlania wszystkich atomów wodoru, wynik powinien być podobny do: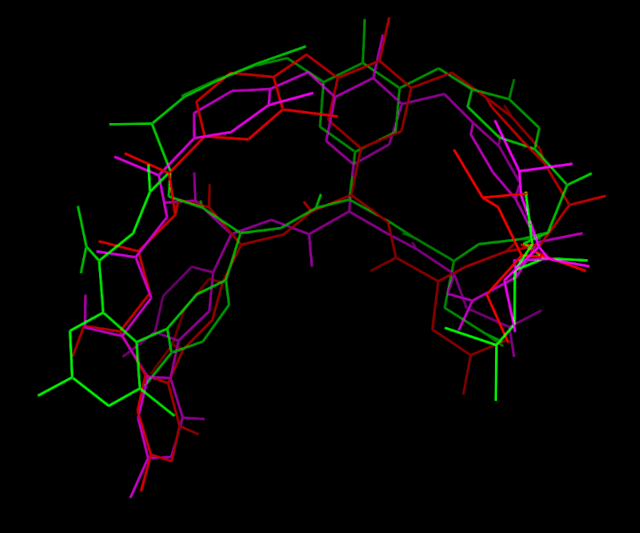 Dyskusja i opis takiego rysunku powinna skupiać się przede wszystkim na zaobserwowanych zmianach w konformacji/różnicach pomiędzy uzyskanymi wynikami MD różnych kompleksów. Na rysunku, powyżej, struktura oznaczona kolorem zielonym odkształciła się najbardziej w obszarze C-końcowym, co pociągnęło za sobą deformację części makrocyklicznej w obszarze reszt 5, 7 i 6 (wszystkie Wasze starty miały tę samą konformację wankomycyny, minimalnie tylko dostosowaną do pentapeptydu).
Dyskusja i opis takiego rysunku powinna skupiać się przede wszystkim na zaobserwowanych zmianach w konformacji/różnicach pomiędzy uzyskanymi wynikami MD różnych kompleksów. Na rysunku, powyżej, struktura oznaczona kolorem zielonym odkształciła się najbardziej w obszarze C-końcowym, co pociągnęło za sobą deformację części makrocyklicznej w obszarze reszt 5, 7 i 6 (wszystkie Wasze starty miały tę samą konformację wankomycyny, minimalnie tylko dostosowaną do pentapeptydu).
- w podobny sposób można przygotować nałożenie samych pentapeptydów ze wszystkich kompleksów
- wskazane byłyby także tabele zawierające zebrane ze wszystkich kompleksów uśrednione, końcowe wartości energii całkowitej, potencjalnej i kinetycznej, odczytane z plików summary_avg.ETOT, summary_avg.EPTOT i summary_avg.EKTOT - z ostatnich linijek (opisanym klatką "1000")
- stabelaryzować także należy skład cząsteczkowy/atomowy poszczególnych układów symulacyjnych (na przykład ile cząsteczek wody było w pudle, ile i jakich przeciwjonów, jakiego rozmiaru/wielkości pudło zostało utworzone i jakie były wymiary pudła symulacyjnego po dynamice; rozmiary pudła są zapisane w ostatniej linijce plików CRD - tuż przed trzeba liczbami "90" - definującymi geometrie pudła)
- no i oczywiście dyskusja i wnioski nt. skuteczności modyfikacji struktury wankomycyny na wiązanie pentapeptydu z wankomycyną i energetykę takich kompleksów.
PD - powrót.