Rafał Ślusarz
Aby ułatwić sobie ocenę tego, co się "wydarzyło" z konformacją kompleksu (jego składników), proponuję:
- złożyć wszystkie trajektorie do jednego pliku, pozbawiając je jednocześnie cząsteczek wody
- dostosować plik z topologią kompleksu
- ocenić (wizualnie; ręcznie) zmiany, jakie miały miejsce podczas MD używając programu PyMOL
trajin 00.crd trajin 01.trj trajin 02.trj trajin 03.trj trajin 04.trj strip :WAT center :1-7 trajout cala_trajektoria.trj run
Można go nazwać np. ptraj_sklejenie.in
Powyższy plik wejściowy usuwa z całej trajektorii wszystkie cząsteczki wody i połączony plik z zapisem przebiegu MD zapisuje pod nazwą cala_trajektoria.trj. W pliku tym struktury są w każdym kroku centrowane, przy czym centrum obrazu stają się reszty od 1 do 7. Jest to działanie dobre dla wszystkich kompleksów, niezależnie od modyfikacji (do centrum jest sprowadzany aglikon wankomycyny, dzięki czemu, podczas oglądania zapisu trajektorii, cząsteczki kompleksu zawsze będą na środku ekranu/okna).
Uruchomienie "sklejenia" (można to zrobić interaktywnie, trwa bardzo krótko):
cpptraj -p plik_topologii.top -i ptraj_sklejenie.in
Plik wejściowy ptraj_sklejenie.in musi być zapisany w katalogu, w którym znajdują się pliki trajektorii poszczególnych przebiegów MD; tam też należy wykonać/uruchomić powyższe polecenie.
Nowej trajektorii musi odpowiadać nowa topologia.
Aby przygotować nową topologię, należy interaktywnie użyć programu rdparm w taki sposób:
rdparm oryginalna_topologia.top
Wchodzimy w ten sposób do interaktywnej edycji pliku TOP.
Akcja, którą chcemy wykonać, to usunięcie cząsteczek wody. Wykonuje się to poleceniem:
stripwater
Program podsumuje stan układu i zapyta nas w tym momencie, czy jesteśmy pewni tego, co chcemy właśnie zrobić. Odpowiadamy twierdząco:
yes
kolejne pytanie będzie dotyczyło tego, ile cząsteczek wody ma pozostać w układzie po usunięciu wody - może trochę przewrotne, ale zwraca uwagę na to, co właśnie chcemy zrobić. Chcemy usunąć wszystkie cząsteczki wody, więc po naszej akcji w układzie tych cząsteczek ma zostać dokładnie zero:
0
Ostatnie pytanie to moment, w którym możemy zdecydować, jak ma się nazywać nowy plik, zawierający zmodyfikowaną topologię (bez wody). Proponuję coś w stylu:
Po tym pozostaje już tylko zakończenia działania programu rdparm:
Dysponujemy teraz parą nowych plików: cala_trajektoria.trj i topologia_bez_wody.top, które należy przesłać do komputera lokalnego i wczytać do analizy do programy PyMOL.
Jeżeli PyMOL nie rozpoznaje formatów plików i nie potrafi załadować (w powyższy sposób) historii dynamiki, to należy my formaty wyspecyfikować rozbudowaną wersją tych samych poleceń:
Słowo calosc, powyżej, to nazwa obiektu, do którego została załadowana najpierw topologia, a potem wszystkie klatki całej trajektorii.
Kolejnym krokiem musi być zmiana wyświetlania załadowanego obiektu.
Dodatkowo, aby wyróżnić sam pentapeptyd, należy go wybrać i wyróżnić. Można to zrobić na kilka sposobów. Najprostszym wydaje się być:
Obiekt (sele) możemy teraz wyświetlić na przykład w postaci pogrubionych drucików:
Teraz możemy kliknąć obok kompleksu (w czarne pole), aby zdjąć zaznaczenie (podświetlenie) wszystkich atomów pentapeptydu.
Otrzymujemy w ten sposób dość czytelną strukturę wankomycyny i pentapeptydu, których wzajemne zachowanie w trakcie MD możemy prześledzić używając automatycznego odtwarzania (przyciski sterujące znajdują się z prawej strony okna głównego PyMOL-a, u dołu) lub korzystając z paska postępu u dołu całej szerokości okna głównego PyMOL-a (mozna "złapać" za ten wskaźnik postępu MD i go przeciągnąć do miejsca, które nas interesuje w historii MD).
W ten sposób możemy dosłownie zobaczyć, co się działo z całym układem w całym przebiegu MD, zatrzymać się w konkretnym miejscu symulacji i sprawdzić konformację lub oddziaływania poszczególnych fragmentów (a także wykonać zrzuty grafiki poszczególnych, szczególnie interesujących stanów kompleksu).
Całe to postępowanie należy przeprowadzić osobno dla każdego symulowanego kompleksu (File > Export image as... > PNG).
PD - powrót.
topologia_bez_wody.top
quit
Aby wczytać całą historię dynamiki do PyMOL-a, należy zacząć od załadowania topologii. Współczesne (nowe) wersje PyMOL-a powinny automatycznie rozpoznawać format ładowanych plików, w takiej sytuacji wystarczy wydać w linii komend tego programu dwa polecenia:
load topologia_bez_wody.top
load_traj cala_trajektoria.trj
load topologia_bez_wody.top, calosc, 1, top
load_traj cala_trajektoria.trj, calosc, 1
Domyślnie PyMOL będzie próbował interpretować i wyświetlać rozpoznane struktury drugorzędowe, co w naszym przypadku prowadzi do wyświetlenia jedynie kilku fragmentów całego kompleksu. Aby wyeliminować ten efekt należy włączyć wyświetlanie WSZYSTKIEGO w postaci linii, wybierając polecenie Show:
S > as > lines
* - jeżeli kliknięcie się nie uda (traficie w wankomycynę, a nie w pentapeptyd, to zaznaczenie można zdjąć klikając gdziekolwiek poza jakąkolwiek strukturą (w czarny, pusty obszar)
Ilustracja do poszczególnych punktów: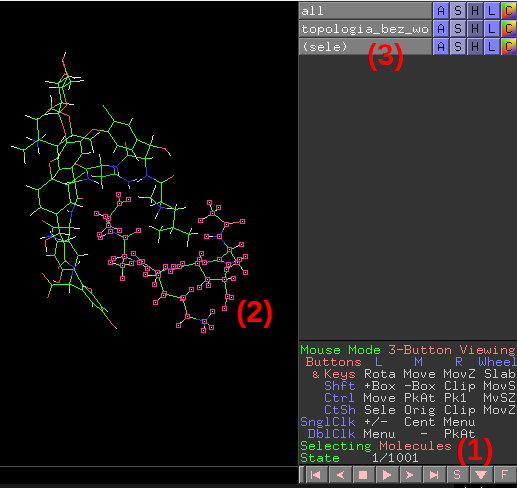
S > as > sticks
Numer wyświetlanej aktualnie klatki z całej trajektorii (zrzutu stanu układu, zapisywanego w naszych symulacjach co 1000 kroków MD) jest wyświetlona w postaci licznika z prawej strony, u dołu okna głównego, w polu State.